Advances in Enzyme Replacement Therapy for Fabry Disease: Improving Patient Outcomes
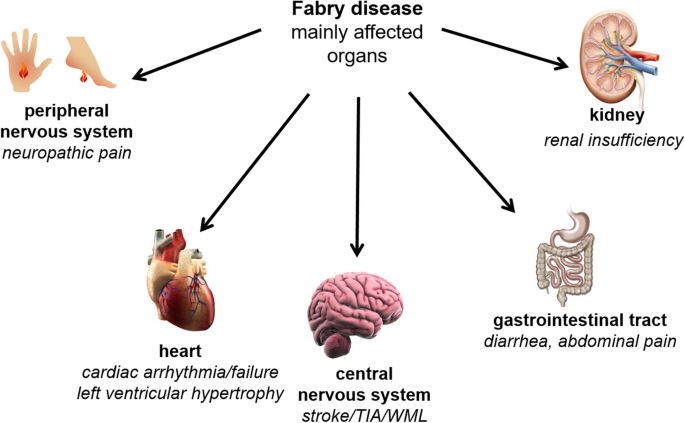
Fabry Disease is a rare genetic disorder characterized by the deficiency or dysfunction of an enzyme called alpha-galactosidase A (α-Gal A), which leads to the accumulation of a specific type of fatty substance called globotriaosylceramide (Gb3) in various organs and tissues. This progressive accumulation results in a wide range of debilitating symptoms, including pain, kidney dysfunction, heart complications, and neurological issues. Over the years, Enzyme Replacement Therapy (ERT) has emerged as a promising treatment avenue for Fabry Disease, offering significant improvements in patient outcomes and quality of life.
ERT involves administering a synthetic form of the missing or dysfunctional enzyme, α-Gal A, intravenously to patients. This therapeutic approach aims to address the root cause of Fabry Disease by providing the enzyme that the body lacks, thereby reducing the accumulation of Gb3 in cells and tissues. While ERT doesn't offer a complete cure, it has demonstrated remarkable potential in alleviating symptoms and slowing disease progression.
The development of ERT for Fabry Disease began in the early 2000s, with the introduction of agalsidase alfa and agalsidase beta, two recombinant forms of α-Gal A. These therapies have been successful in reducing Gb3 levels and improving organ function in many patients. However, the effectiveness of ERT can vary depending on factors such as the severity of the disease, patient age, and the presence of organ damage at the time of treatment initiation.
In recent years, research has focused on refining ERT approaches to enhance its efficacy and improve patient outcomes. One significant advancement is the development of next-generation enzyme replacement therapies, including pegunigalsidase alfa and velaglucerase alfa. These therapies are designed to have improved pharmacokinetic properties, which can lead to longer-lasting effects and potentially reduce the frequency of infusions. This innovation not only increases convenience for patients but also offers the potential for more consistent enzyme levels, which could translate to better disease control.
Moreover, personalized medicine approaches are being explored to tailor ERT regimens to individual patients. Genetic and clinical factors are considered to optimize dosage, treatment frequency, and monitoring strategies. This patient-centric approach aims to maximize the benefits of ERT while minimizing potential side effects and complications, further contributing to improved patient outcomes.
One of the challenges of ERT is its limited ability to address certain aspects of Fabry Disease, particularly neurological symptoms. The blood-brain barrier can hinder the delivery of therapeutic enzymes to the central nervous system. However, ongoing research is exploring strategies to enhance the brain's uptake of α-Gal A and improve treatment efficacy for neurological complications.
In conclusion, advances in Enzyme Replacement Therapy for Fabry Disease are providing significant benefits to patients by improving their quality of life and slowing disease progression. The development of next-generation therapies, personalized treatment strategies, and ongoing research into addressing neurological symptoms all contribute to the evolving landscape of Fabry Disease treatment. While ERT has undoubtedly revolutionized the management of this rare genetic disorder, continued research and innovation remain critical to further enhance its effectiveness and broaden its impact on patient outcomes.
Top of Form